
目次
午前
データ整理
午後
データ整理
会議(学外)
データ整理
サンプル間の遺伝距離をRで計算する方法に試行錯誤をしたのでメモ。
塩基配列データに基づき遺伝距離を計算するだけなら「ape::dist.dna()」を使えば良い。
library(ape)
dna_name <- read.dna(FASTAデータ)
dist <- dist.dna(dna_name)
dist
ここからが苦戦した。このデータをRで使用しやすい縦型のデータにしたい。
一発でやる方法がありそうなのだが、本日見つけた方法は、まずマトリックス型に変形する。
dis_matrix <- as.matrix(dist)
dis_matrix
このうち重複している左下(or右上)と同じサンプルの組み合わせになっている対角部分を削除したい。
これには「base::lower.tri()」が使える。「diag=T」にすることで対角部分にもNAが入る。
dis_matrix[lower.tri(dis_matrix, diag = T)] <- NA
dis_matrix
そして、本日、最も大きな発見は、このマトリックス型を一撃で縦長データにする関数「reshape2::melt()」を見つけたこと!
library(reshape2)
dis_long <- melt(dis_matrix)
dis_long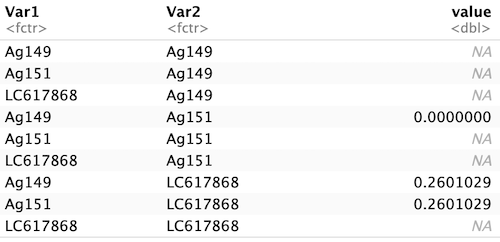
ここまでくればvalue列からNAを除くだけ!
dis_select <- dis_long %>%
drop_na(value)
dis_select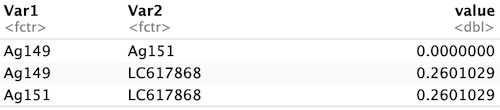
遺伝距離を計算するだけならMEGAを使えば簡単にできる。しかもグループ化もできる。
ただ、そのあと色々と計算する場合にはR上で縦長データにしておいた方が便利、、、な気がして頑張ってみた。そこまで必要なかったかも。
【参考文献】